SUPPORT
Provide support by our committed team of families to newly diagnosed individuals, and patients diagnosed with Arginase 1 Deficiency.
RESEARCH
Dedicated to education and research in support of treatment and ultimately a cure for Arginase 1 Deficiency.
EDUCATION
Promote early diagnosis, increase awareness and recognition of Arginase 1 Deficiency among healthcare professionals.

Welcome
Welcome to the patient advocacy website for support and information regarding Arginase 1 Deficiency (ARG1-D). We are glad you found us. Our community is made up of parents, family members, friends and caregivers who have also faced this diagnosis and understand the range of emotions and questions you may have. We are here to provide information on ARG1-D, resources to guide you on this journey and perhaps most importantly, we are here to let you know that you are not alone. We hope you find this website a valuable resource as you learn more about ARG1-D, and that you reach out to us should you have additional questions or needs.
Video introduction to Arginase 1 Deficiency
Video made possible by the support of Aeglea Biotherapeutics.
Dr. Emily Shelkowitz
What is Arginase 1 Deficiency?
Arginase 1 Deficiency (ARG1-D) is a rare, progressive, inherited disease that typically presents in childhood. It is an inherited disorder that affects children, teens and adults, and may significantly impact a patient’s health over time.
ARG1-D is characterized by complete or partial lack of the enzyme arginase in the liver and red blood cells. Arginase is one of six enzymes that play a role in the breakdown and removal of nitrogen from the body, a process known as the urea cycle. The lack of the arginase enzyme results in excessive accumulation of nitrogen, in the form of ammonia (hyperammonemia), in the blood and arginine (hyperarginemia) in the blood and cerebrospinal fluid. Untreated children may exhibit seizures, spasticity, short stature and intellectual disability. Arginase 1 deficiency is inherited as an autosomal recessive genetic disorder.
Those affected with ARG1-D experience persistently high levels of an amino acid called arginine, which leads to muscle tightness (spasticity), missed developmental milestones, intellectual disability, seizures and early death.
ARG1-D Quick Facts
- Caused by the body’s inability to break down arginine
- High arginine levels impact a person’s ability to function
- Symptoms typically appear between 2 to 4 years of age and persist through adulthood
- In rare cases symptoms can present after puberty
Behind the Mystery Segment: Arginase 1 Deficiency
Dr. Emily Shelkowitz
Arginase 1 Deficiency Symptoms and Diagnosis
ARG1-D is 1 of 8 urea cycle disorders (UCDs), but symptoms associated with arginase 1 deficiency differ. Unlike other urea cycle disorders, infants with ARG1-D typically do not experience severe hyperammonemia or hyperammonemic coma. In fact, most infants with ARG1-D do not exhibit any symptoms during the first few months to a year of life.
As children diagnosed with ARG1-D approach one to three years of age, they may experience a lag in growth, may walk on their toes and develop progressive stiffness and lack of control of voluntary movements of the legs (spastic diplegia). Cognitive development slows or stops, and children likely will develop severe spasticity, an inability to walk, loss of bowel and bladder control and severe intellectual disability. In rare cases, symptoms can present after puberty. ARG1-D is a heterogeneous disease, meaning that symptoms and severity will vary. However, almost all affected children have growth deficiency and many also experience seizures.
There are no approved treatments to address the root cause of ARG1-D, but there is research underway. This program provides hope to our community, and we will continue to raise our voices and advocate for more attention to this disease. When more information becomes available about this Phase 3 study, we will share it with you.
Additionally, if you suspect your child or someone you know may have symptoms of ARG1-D, diagnostic testing if available for adults and children free of cost.

Basics of Arginase 1 Deficiency
- Tripping, stumbling, falling, toe-walking
- Muscle tightness (spasticity)
- Missed developmental milestones
- Protein Adversion
- Intellectual delays
- Seizures
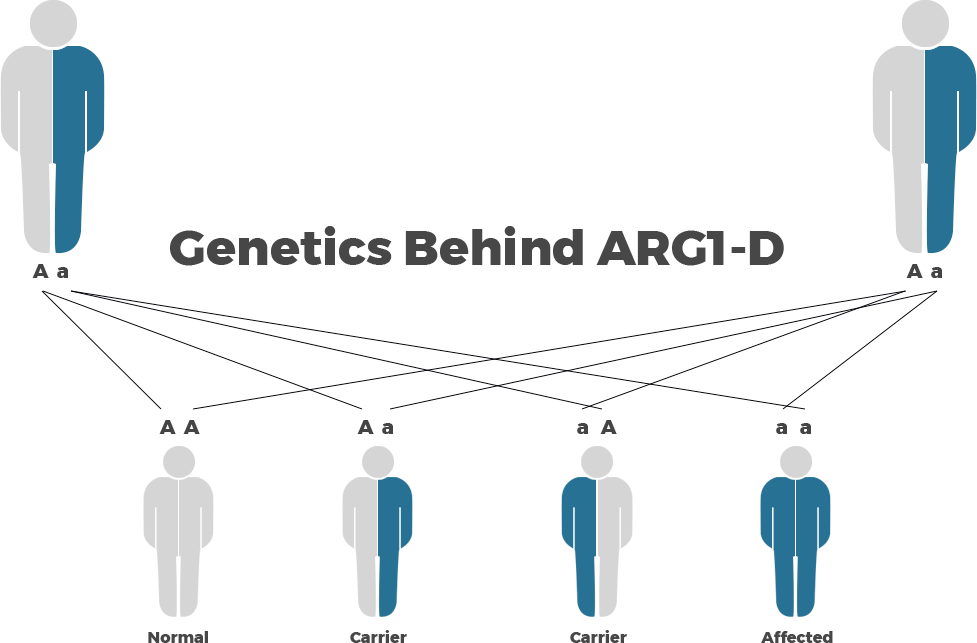
The Genetics Behind Arginase 1 Deficiency
Arginase 1 Deficiency is inherited is an autosomal recessive genetic disorder and is caused by mutations in the ARG1 gene. Mutations in the ARG1 gene result in production of an abnormal arginase enzyme.
Autosomal recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Affected Population
- Arginase 1 Deficiency is among the rarest of rare diseases. It has been estimated to occur in approximately 1 in 300,000-1,000,000 births.
Diagnosing Arginase 1 Deficiency
Most affected infants are now identified at birth through newborn screening. Unfortunately, not all states automatically test for ARG1-D and infants may not exhibit symptoms immediately at birth. While important, newborn screening has limited effectiveness due to low levels of plasma arginine in newborns and inconsistent testing standards. People with ARG1-D may be misdiagnosed with other conditions such as cerebral palsy or hereditary spastic paraplegia. Plasma amino acid panels and genetic tests are considered to be more reliable and are readily available. Early detection of ARG1-D is critical and can help your child get the treatment he/she needs. ARG1-D can be diagnosed easily, and for those in need these tests are accessible free of cost.
Testing is simple and
can be a mix of the following
- Newborn screening is done routinely in some states and by request in other states
- Blood (plasma) test for amino acids that show arginine levels
- Genetic testing
Treatment for Arginase 1 Deficiency
There are currently no approved treatments to specifically address the underlying cause of ARG1-D, but late-stage research is underway that gives us hope. We will continue to advocate as a community for more awareness, focus and treatment options for those living with this rare but devastating disease.
There are important steps to support a patient’s well-being. A metabolic team of specialists including a nutritionist/dietician should be part of any patient’s care team, with a focus on reducing plasma ammonia and arginine concentration, preventing excess ammonia from being formed and reducing the amount of nitrogen in the diet.
Dietary restrictions for individuals with ARG1-D are very important and aimed at limiting the amount of arginine and protein intake. Individuals with ARG1-D are placed on a low-protein, arginine-restricted diet supplemented by essential amino acids.
It is also important to help reduce the amount of plasma ammonia concentration. Some people may be prescribed oral nitrogen scavenger drugs sodium phenylbutyrate and sodium benzoate. Ammonul can be administered intravenously to treat the excess ammonia in the blood. In extreme cases dialysis may be used.
Seizures are treated with a variety of medications such as phenobarbital or carbamazepine. Valproic acid for the treatment of seizures should be avoided as it can increase blood ammonia levels. Always talk to your doctor about the pros and cons of the medications that are available and decide what is right for you or for you and your child.
Affected individuals should receive periodic blood tests to determine the levels of ammonia and arginine in the blood and to be sure that liver function is not impaired. Excessive levels of ammonia or arginine should be promptly treated.
Genetic counseling is recommended for affected individuals and their families, and as always check with the patient’s doctor before beginning any treatment plan.

Low-protein,
arginine-restricted diet
- Dietary restrictions in individuals with Arginase 1 Deficiency are aimed at limiting the amount of arginine and protein intake. Patient’s with Arginase 1 Deficiency are placed on a low-protein, arginine-restricted diet supplemented by essential amino acids.